
« Avec le soutien institutionnel du laboratoire ADVICENNE »
Dossier ATRd _ Partie 1
Pr Olivia Gillion Boyer : Service de néphrologie pédiatrique, centre de référence MARHEA, Institut Imagine, Hôpital Necker Enfants Malades, APHP. Centre, Université Paris Cité, Paris France. 15 Avr 2024.
Les acidoses tubulaires rénales (ATR) sont un groupe de pathologies caractérisées par un défaut de réabsorption des bicarbonates et/ou de sécrétion d’acides non volatiles par les différents segments du tubule rénal, conduisant à un déséquilibre acido-basique. Elles doivent être évoquées devant une acidose métabolique hyperchlorémique à trou anionique plasmatique normal (Na+ + K+ − Cl− − HCO3− = 16 ± 4 mEq/L) et trou anionique urinaire positif (Na+ + K+ − Cl−) (Figure 1).
On en distingue quatre types : l’ATR distale (ATRd, ou de type 1), caractérisée par un défaut de sécrétion urinaire de protons H + par le tubule distal et le tubule connecteur ; l’ATR proximale (ou de type 2), secondaire à un défaut de réabsorption des bicarbonates (HCO3−) par le tubule proximal, l’ATR de type 3 ou mixte ; et l’ATR de type 4 due à un déficit en aldostérone ou à une résistance des tubules rénaux à l’aldostérone.
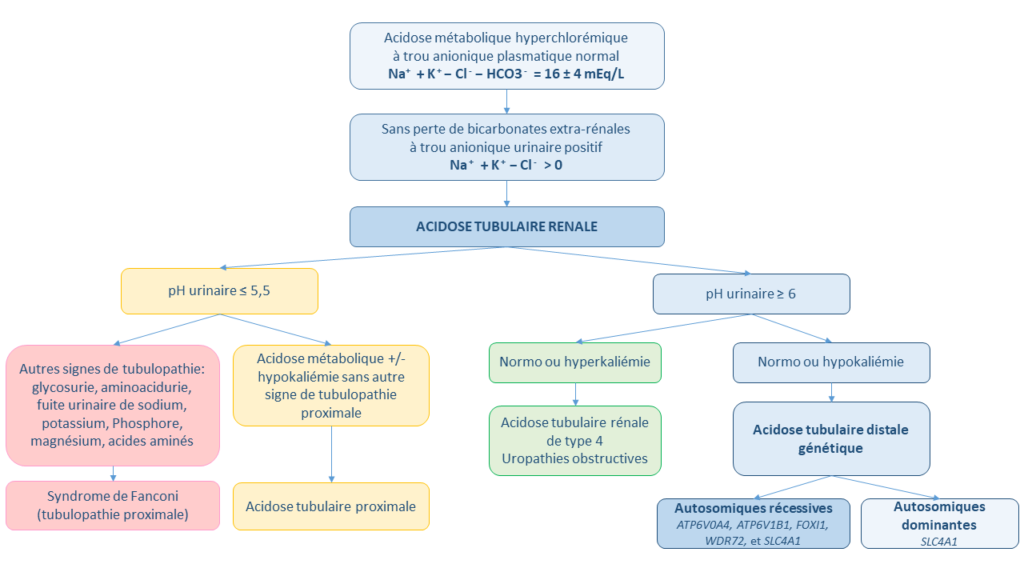
L’ATRd est une pathologie rare, qui peut être héréditaire ou secondaire à des maladies auto-immunes, à la drépanocytose, à des uropathies obstructives chroniques ou à la transplantation rénale.
Cette revue porte sur l’ATRd (ATR de type 1) génétique. Il s’agit de la forme la plus fréquente d’ATR chez l’enfant. Elle résulte d’une incapacité des cellules intercalaires α du tubule distal et du tubule connecteur à acidifier les urines pour éliminer les acides produits par l’alimentation. Ainsi, le pH urinaire ne s’abaisse pas malgré l’acidose (pHu ≥ 6).
Clinique et corrélation phénotype-génotype.
L’ATRd génétique est causée par des variants pathogènes de gènes impliqués dans l’équilibre acido-basique du rein : ATP6V0A4, ATP6V1B1, FOXI1, WDR72, et SLC4A1. Elle peut être de transmission autosomique récessive (AR) fréquemment diagnostiquée chez le nourrisson et le jeune enfant, ou autosomique dominante (AD) de révélation plus tardive chez l’adolescent et l’adulte jeune. L’étude génétique est fondamentale pour confirmer le diagnostic, orienter la prise en charge et le conseil génétique.
1) ATRd autosomique récessive (AR)
L’ATRd AR se présente généralement dans la petite enfance avec un retard de croissance staturo-pondérale, des vomissements, une constipation, une hypotonie, et des épisodes de déshydratation. Elle est associée à une hypokaliémie (par stimulation du système rénine angiotensine aldostérone), une hypercalciurie et une hypocitraturie (par stimulation de la réabsorption proximale du citrate couplée au sodium et mobilisation du tampon osseux) entraînant lithiase, néphrocalcinose, retard de croissance voire déminéralisation osseuse (rachitisme, ostéomalacie). L’hypokaliémie peut induire des arythmies cardiaques, une faiblesse musculaire, une paralysie, voire le décès. L’insuffisance rénale chronique, en particulier les stades 2 et 3, est plus fréquente et survient plus tôt que dans la population générale.
L’ATRd AR avec ou sans surdité de perception est généralement causée par des mutations bialléliques (homozygotes ou hétérozygotes composites) dans les gènes ATP6V0A4 ou ATP6V1B1, codant respectivement pour les sous-unités A4 et B1 de la pompe H+ATPase exprimées dans les cellules intercalaires α du tubule distal et du tubule connecteur du néphron (unité fonctionnelle du rein), et exprimées au niveau de la cochlée appartenant à l’oreille interne. La surdité de perception bilatérale progressive et irréversible, de sévérité très variable, peut être présente au diagnostic ou se manifester plus tardivement. Elle peut s’associer à une dilatation des aqueducs vestibulaires. Les autres rares causes génétiques sont des mutations de FOXI1 entraînant une ATRd AR associée à une perte auditive précoce, et des mutations de WDR72, qui ont été décrites chez 2 familles avec une forme plus légère d’ATRd et des anomalies dentaires. Enfin, des mutations du gène SLC4A1 sont associées ATRd AR si elles sont bialléliques ou AD si elles sont hétérozygotes (monoalléliques, cf infra).
2) ATRd autosomique dominante (AD)
L’ATRd AD, est due à des mutations hétérozygotes (monoalléliques) du gène SLC4A1 codant pour l’échangeur chlore/bicarbonate (AE1) exprimé par les cellules intercalaires α du tubule distal et du tubule connecteur du néphron, et sur la membrane des globules rouges. L’ATRd AD est de révélation plus tardive chez l’adolescent ou le jeune adulte, en général découverte lors d’un bilan de lithiase, de néphrocalcinose ou d’ostéoporose avec hypercalciurie et hypocitraturie. L’acidose hyperchlorémique et l’hypokaliémie sont plus modérées, voire absentes. L’acidose sera révélée dans ce cas par un test de charge acide.
Certains patients porteurs de variants spécifiques de SLCA1 présentent également une anémie hémolytique de type sphérocytose ou ovalocytose, plus fréquente dans la population du Sud-Est asiatique.
Traitement
Des recommandations de prise en charge de l’ATRd ont été établies par des experts de l’European Society for Paediatric Nephrology et de l’European Rare Kidney Disease Reference Network et sont résumées ici. Le traitement dépend des symptômes et de leur gravité. Le but est de maintenir des taux plasmatiques de bicarbonate, de chlore et de potassium, ainsi qu’une calciurie dans les normes correspondant à l’âge. Cela permet de normaliser la croissance chez les enfants et la minéralisation osseuse, et de prévenir la progression de la néphrocalcinose et la formation de calculs.
1) Traitement alcalinisant
Choix du traitement
Le traitement repose sur une supplémentation alcaline dont il existe de nombreuses formes (bicarbonate ou citrate de potassium, de sodium ou de magnésium), liquides ou solides, de courte ou longue durée d’action. Le choix dépend :
- de la disponibilité,
- du coût,
- du goût,
- de la facilité d’administration,
- de la tolérance,
- de la présence ou non d’une hypokaliémie ou d’une hyperkaliémie, notamment chez l’insuffisant rénal,
- de la forme galénique souhaitée (libération immédiate ou libération prolongée),
- du nombre de prises (préparation magistrale : 4 prises par jour ou plus pour couvrir le nycthémère ; forme à libération prolongée : 2 prises par jour).
Dose initiale
La posologie initiale dépend de l’âge et du mode de révélation. Chez les nourrissons présentant une acidose marquée (HCO3– < 15 mmol/L), une dose initiale d’environ 5 mEq/kg/jour de traitement alcalinisant est recommandée. On effectue une titration en surveillant les taux résiduels toutes les 24-48 heures pour ajuster la dose, jusqu’à l’obtention d’une bicarbonatémie normale et stable. En cas de dépistage familial avant le stade d’acidose, une dose initiale de 1 à 2 mEq/kg/jour est en général suffisante. Pour les plus grands enfants la dose initiale recommandée est de 2 à 3 mEq/kg/jour. Enfin, chez les adultes, une dose initiale de 60 mEq/jour est raisonnable avec une augmentation de 15-20 mEq toutes les 1 à 2 semaines jusqu’à ce que les paramètres biochimiques se soient normalisés.
2) Supplémentation potassique
Une supplémentation potassique est recommandée chez les patients présentant une hypokaliémie persistante malgré un bon contrôle de l’acidose et une fonction rénale préservée. L’utilisation de citrate ou de bicarbonate de potassium présente l’avantage d’alcaliniser et de supplémenter en potassium simultanément.
Chez l’insuffisant rénal, la supplémentation potassique risque d’induire une hyperkaliémie et doit être prescrite avec précaution et sous surveillance rapprochée par un spécialiste. L’utilisation de diurétiques thiazidiques n’est pas recommandée en routine.
3) Traitement des complications et des atteintes extra-rénales
Pour les ATRd récessives liées à des variants des gènes ATP6V1B1, ATP6V0A4 ou FOXI1, un dépistage auditif précoce (avant l’âge de 30 mois) est recommandé. Les patients présentant une surdité de perception doivent faire l’objet d’une prise en charge spécialisée rapide.
Chez les adultes, la mesure de la densité minérale osseuse par ostéodensitométrie tous les 2 ou 3 ans peut être utile pour évaluer le risque fracturaire et adapter le traitement. Cet examen n’est pas recommandé systématiquement chez les enfants du fait des difficultés d’interprétation et de la grande variabilité dans le temps.
Comme la plupart des enfants ont une croissance normale avec un contrôle adéquat de l’acidose, un traitement par hormone de croissance n’est en général pas recommandé en dehors de l’insuffisance rénale chronique. Un suivi nutritionnel par un ou une diététicien(ne) spécialisé(e) peut être utile pour réduire la charge acide du régime alimentaire (protéines animales, aliments à indice PRAL positif) en maintenant un état nutritionnel adéquat.
Surveillance
Il est recommandé de surveiller la croissance, les paramètres biochimiques (ionogramme sanguin, calciurie) et l’échographie des voies urinaires à intervalles réguliers à la recherche d’une lithiase et/ou néphrocalcinose.
REFERENCES :
- Trepiccione F, Walsh SB, Ariceta G, Boyer O, Emma F, Camilla R, Ferraro PM, Haffner D, Konrad M, Levtchenko E, Lopez-Garcia SC, Santos F, Stabouli S, Szczepanska M, Tasic V, Topaloglu R, Vargas-Poussou R, Wlodkowski T, Bockenhauer D. Distal renal tubular acidosis: ERKNet/ESPN clinical practice points. Nephrol Dial Transplant. 2021 Aug 27;36(9):1585-1596.
- Giglio S, Montini G, Trepiccione F, Gambaro G, Emma F. Distal renal tubular acidosis: a systematic approach from diagnosis to treatment. J Nephrol. 2021 Dec;34(6):2073-2083.
- Hureaux M, Ashton E, Dahan K, Houillier P, Blanchard A, Cormier C, Koumakis E, Iancu D, Belge H, Hilbert P, Rotthier A, Del Favero J, Schaefer F, Kleta R, Bockenhauer D, Jeunemaitre X, Devuyst O, Walsh SB, Vargas-Poussou R. High-throughput sequencing contributes to the diagnosis of tubulopathies and familial hypercalcemia hypocalciuria in adults. Kidney Int. 2019 Dec;96(6):1408-1416.
- Palazzo V, Provenzano A, Becherucci F, Sansavini G, Mazzinghi B, Orlandini V, Giunti L, Roperto RM, Pantaleo M, Artuso R, Andreucci E, Bargiacchi S, Traficante G, Stagi S, Murer L, Benetti E, Emma F, Giordano M, Rivieri F, Colussi G, Penco S, Manfredini E, Caruso MR, Garavelli L, Andrulli S, Vergine G, Miglietti N, Mancini E, Malaventura C, Percesepe A, Grosso E, Materassi M, Romagnani P, Giglio S. The genetic and clinical spectrum of a large cohort of patients with distal renal tubular acidosis. Kidney Int. 2017 May;91(5):1243-1255.
- Lopez-Garcia SC, Emma F, Walsh SB, Fila M, Hooman N, Zaniew M, Bertholet-Thomas A, Colussi G, Burgmaier K, Levtchenko E, Sharma J, Singhal J, Soliman NA, Ariceta G, Basu B, Murer L, Tasic V, Tsygin A, Decramer S, Gil-Peña H, Koster-Kamphuis L, La Scola C, Gellermann J, Konrad M, Lilien M, Francisco T, Tramma D, Trnka P, Yüksel S, Caruso MR, Chromek M, Ekinci Z, Gambaro G, Kari JA, König J, Taroni F, Thumfart J, Trepiccione F, Winding L, Wühl E, Ağbaş A, Belkevich A, Vargas-Poussou R, Blanchard A, Conti G, Boyer O, Dursun I, Pınarbaşı AS, Melek E, Miglinas M, Novo R, Mallett A, Milosevic D, Szczepanska M, Wente S, Cheong HI, Sinha R, Gucev Z, Dufek S, Iancu D; European dRTA Consortium; Kleta R, Schaefer F, Bockenhauer D. Treatment and long-term outcome in primary distal renal tubular acidosis. Nephrol Dial Transplant. 2019 Jun 1;34(6):981-991.
- Bertholet-Thomas A, Guittet C, Manso-Silván MA, Castang A, Baudouin V, Cailliez M, Di Maio M, Gillion-Boyer O, Golubovic E, Harambat J, Klein A, Knebelmann B, Nobili F, Novo R, Podracka L, Roussey-Kesler G, Stylianou C, Granier LA. Correction to: Efficacy and safety of an innovative prolonged-release combination drug in patients with distal renal tubular acidosis: an open-label comparative trial versus standard of care treatments. Pediatr Nephrol. 2021 Jan;36(1):215.
- Boyer O, Manso-Silván MA, Joukoff S, Berthaud R, Guittet C. Improved growth of a child with primary distal renal tubular acidosis after switching from a conventional alkalizing treatment to a new prolonged-release formulation containing potassium citrate and potassium bicarbonate: lessons for the clinical nephrologist. J Nephrol. 2022 Nov;35(8):2119-2122.
- Acidose.pdf (filiereorkid.com)











